
https://doi.org/10.1140/epjb/e2011-20094-1
PPP Hamiltonian for polar polycyclic aromatic hydrocarbons
1
Departamento de Química Física, Unidad Asociada del CSIC and
Instituto Universitario de Materiales, Universidad
de Alicante, San Vicente del Raspeig, 03690 Alicante, Spain
2
Departamento de Química Orgánica and
Instituto Universitario de Síntesis Orgánica, Universidad
de Alicante, San Vicente del Raspeig, 03690 Alicante, Spain
3
Departamento de Teoría y Simulación de Materiales, Instituto de Ciencia de Materiales de Madrid (CSIC), Cantoblanco, 28049 Madrid, Spain
4
Departamento de Física Aplicada, Unidad Asociada del
CSIC and Instituto Universitario de Materiales, Universidad de Alicante, San Vicente del Raspeig, 03690 Alicante, Spain
Corresponding author: a chiappe@ua.es
Received:
4
February
2011
Revised:
1
April
2011
Published online:
25
May
2011
Total energies of charged states and configurations of different spin multiplicity of two polar non-alternant polycyclic aromatic
hdrocarbons (PAH), namely, pentaheptafulvalene and azulene, calculated by means of a Multi-Configurational (MCSCF) method which includes correlation only amongst π orbitals, have been fitted by
exact solutions of the Pariser-Parr-Pople (PPP)
and the Hubbard
Hamiltonians for π electrons. As both molecules are planar, such an approach is in principle feasible. As found in our previous analysis of PAH, PPP fittings
are significantly better than those attained with the Hubbard Hamiltonian. In addition, parameters for the Hubbard Hamiltonian are around twice those derived for the PPP model, indicating that parameters are not model independent. Fitted PPP parameters are close to those derived from a similar study of the PAH 2, 5, 8-trihydrogenated phenalene and those originally proposed by Pariser et al.
providing further support to a wide applicability of the fitted parameters. Fittings obtained for a MCSCF method that also includes 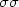 and
and  correlations (MCSCF/MP2) are slightly less accurate giving an on-site repulsion 10–15% smaller. The accuracy of the fittings further diminishes when parameters are derived from energies obtained by means of a DFT method (B3LYP) with an additional decrease in U of 5–25%. In the latter two cases, parameters have to be considered as effective, accounting for effects of σ orbitals not explicitly included in the model Hamiltonians.
Electron affinities, ionization energies and dipole moments, calculated by means of the model Hamiltonians, are compared to those derived from DFT and ab initio methods and, whenever available, to experimental data.
correlations (MCSCF/MP2) are slightly less accurate giving an on-site repulsion 10–15% smaller. The accuracy of the fittings further diminishes when parameters are derived from energies obtained by means of a DFT method (B3LYP) with an additional decrease in U of 5–25%. In the latter two cases, parameters have to be considered as effective, accounting for effects of σ orbitals not explicitly included in the model Hamiltonians.
Electron affinities, ionization energies and dipole moments, calculated by means of the model Hamiltonians, are compared to those derived from DFT and ab initio methods and, whenever available, to experimental data.
© EDP Sciences, Società Italiana di Fisica, Springer-Verlag, 2011


